公司最近在折腾欧盟QP认证事宜,结合之前商业化生产经验,与研发质量体系的搭建,再加上我重新部分资料,详细规定如下(先从上位法EC指令开始,再到第四卷的相关附录13,附录16),见“具体问题”。
前提条件:目前产品在临床阶段(新适应症),生产地址在国内,申办方也在国内,暂时还没有到上市,需要去欧盟找代理的时候),临床阶段的QP和商业化的QP,无太大区别,都要进行QP认证QP放行,只是临床阶段多了一个申办者放行,另外放行之前参考“产品质量标准文件”(大白话就是:产品档案文件,类似于国内的附录13,临床药品质量档案文件)。
具体问题:第三国进口,就指中国,且还没到欧盟上市,需要在欧盟设立仓库和实验室(或者租赁外包,委托的)?
个人理解:是需要,但是几家不同的CDMO,以及找的不同咨询公司(做QP认证)看法不一样,有的认为需要,有的认为不需要,理解需要因为如下原话(1:附录16:识林截图的附录16原话,并核对了英文描述关于从欧盟以外第三方生产,考虑存储和运输,以及必要的定性、定量检验、2:附录13 原话关于临床药品在第三国生产放行,欧盟有部分互认,但中国不属于互认协议中的国家,故此在中国不存在豁免:,大家很多都是抓住这两个截图去歧义理解),但是下面第三张是上位法,即3:“Article 51 of Directive 2001/83/EC,”的article 51,免于系列检验的前提是“成员国完成了上述管控”,但“中国”不属于成员国,不像欧美等部分国家之间签订了MRA互认协议。故此不存在“豁免事宜”。
总结:即在上位法article 51的指导下,结合附录13临床试验用药的生产和附录16药品质量受权人签发证书和放行批产品,这三个综合评判,是需要,大家的看法呢?现在还在不断地咨询新的咨询公司。

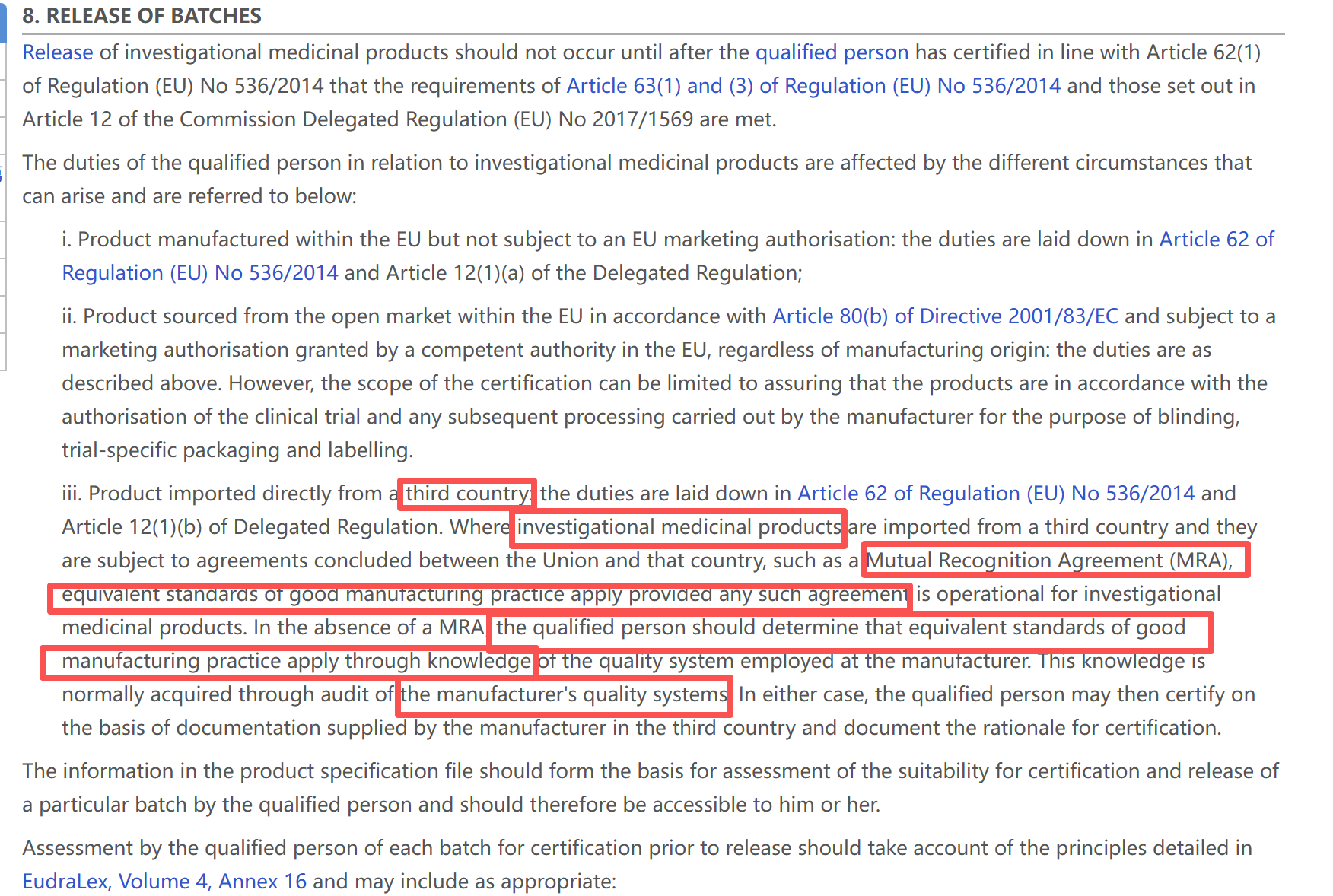

Directive 2001/83/EC (Article 51),其约束的主体是商业化上市药品 。
但是,目前你们的产品处于临床阶段。
临床试验用药 (IMP) 在欧盟不适用 Directive 2001/83/EC,它的最高上位法是 《临床试验法规》 Regulation (EU) No 536/2014 (CTR),以及专门针对IMP的 Delegated Regulation (EU) 2017/1569。
1. 欧盟境内是否需要仓库?
必须需要(必须有物理存放地和进口实体)。
要在欧盟开展临床并进口药物,必须有一个位于欧盟境内的实体持有 MIA(IMP)(临床试验用药生产/进口许可证)。 产品从中国发往欧盟后,物理上必须先清关进入一个符合欧盟GMP/GDP要求的仓库。此时产品处于法定隔离待行状态(Quarantine)。负责放行的欧盟QP必须绑定在这个MIA(IMP)持证实体下。因此,你们必须在欧盟当地租赁一个有资质的仓储中心或委托有MIA(IMP)的代理公司来接收和存放货物,完成法定意义上的申办者放行和QP放行。
2. 欧盟境内是否需要实验室(复检)?
法规层面“不需要强制复检”,但实操层面取决于QP的个人风险偏好。
法规的真实要求: 根据 Regulation (EU) No 536/2014 第63条和 Delegated Regulation 2017/1569,对于从第三国(中国)进口的 IMP,法律并没有像商业化产品那样,强制要求在欧盟境内重新进行全项定量和定性检验。 法规要求的是:QP必须确保该批次产品的生产和检验符合“至少等同于欧盟GMP的标准”,并符合临床试验许可(CTA)和产品标准文件(PSF)。 这意味着,只要欧盟的QP能够通过对中国生产商的现场审计(确认其质量体系等同于欧盟GMP),并审核中国的生产记录和COA,QP在法理上完全可以直接采信中国实验室的检验结果进行放行,不需要在欧盟额外设实验室重检。
①实验室的这个,确实,“(EU) 2017/1569人用临床试验用药品生产质量管理规范(GMP)的原则、指南以及检查相关安排”,后来查了,确实有原话,不强制要求复检。
②仓库的那个事,有道理,不管法规有没有说,总不能随便找个仓库放,后来也查了另外一份文件“依据药物临床试验质量管理规范(GCP)与药品生产质量管理规范(GMP),申办方在人用临床试验用药品储运与运输环节的责任指南Guideline on the responsibilities of the sponsor with regard to handling and shipping of investigational medicinal products for human use in accordance with Good Clinical Practice and Good Manufacturing Practice”明确规定临床期间,GCP和GMP之间的衔接,
这{{threadTextType}}正{{isAdminText}}
为帮助审核人员更快处理,请填写举报原因:
为帮助审核人员更快处理,请填写举报原因: